About CAN-IMMUNE Database
Project Overview
CAN-IMMUNE Contact
| Role | Name | Position | Affiliation | |
|---|---|---|---|---|
| Supervisor | Dr Chen Li | Research Fellow | Monash University | chen.li@monash.edu |
| Developer | Sanjay Krishna | Research Assistant | Monash University | sanjay.krishna@monash.edu |
For questions, bug reports, or collaboration inquiries regarding the CAN-IMMUNE database, please contact us via email.
Project Workflow
-
Mutation Data Collection
Multi-source cancer mutation curation

Mutation data collected from three primary sources:
- COSMIC v100: 4,952,684 missense substitutions from 1,460 cancer cell lines
- CCLE: 968,457 cancer-specific missense substitutions from WES data
- Published Literature: 806 mutations from 86 additional cell lines
Total: 6,721,816 cancer-specific mutations across 33 cancer types
-
Mutation Validation & Cross-referencing
Quality control and verification
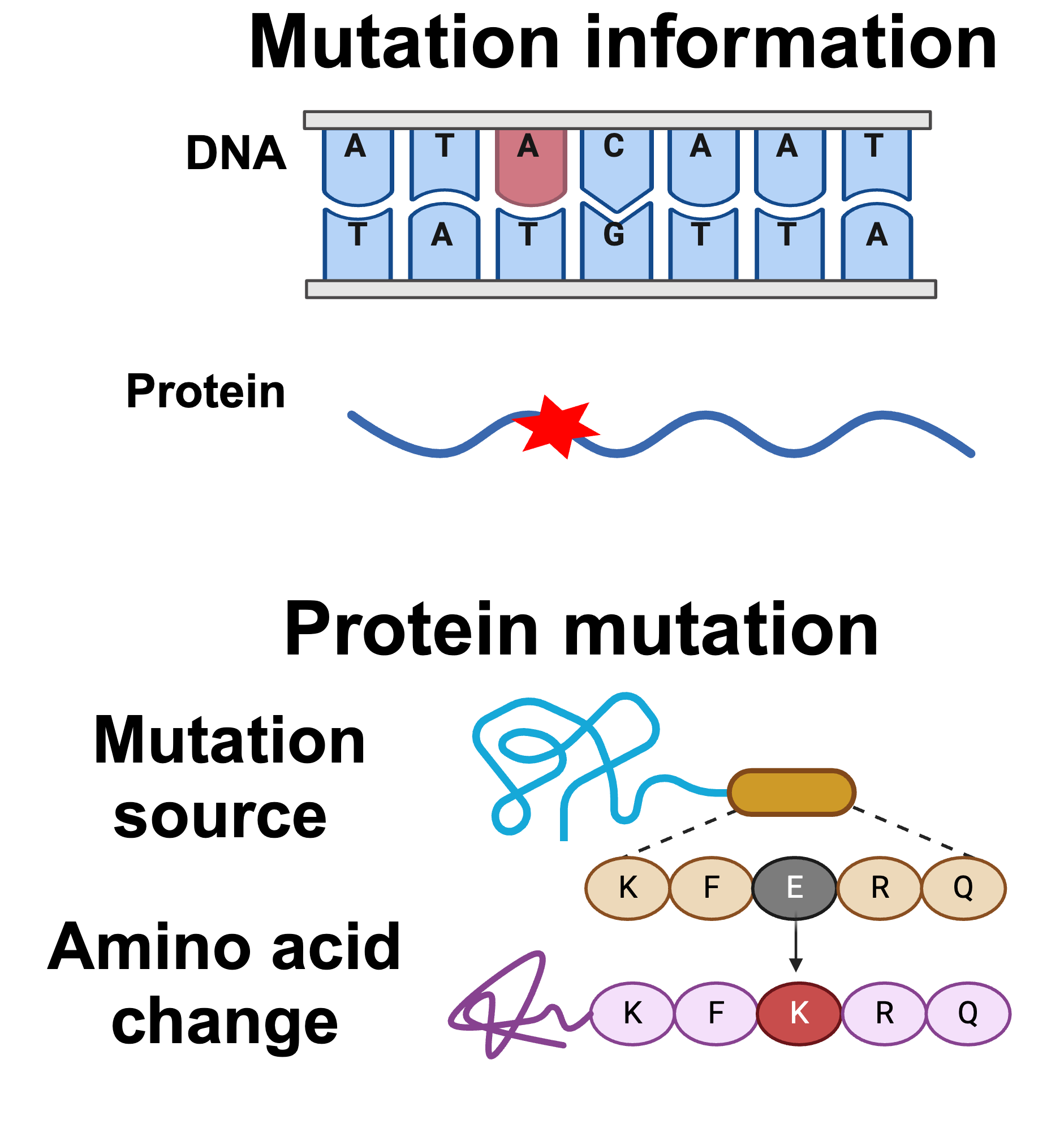
Mutations cross-referenced with established protein databases:
- RefSeq Database (GRCh38): Gene names and positions
- UniProt Database: Amino acid sequences and annotations
Validation ensures accuracy of mutation annotations including gene location, amino acid changes, and transcript IDs
-
Mutant Peptide Library Generation
Creating searchable libraries
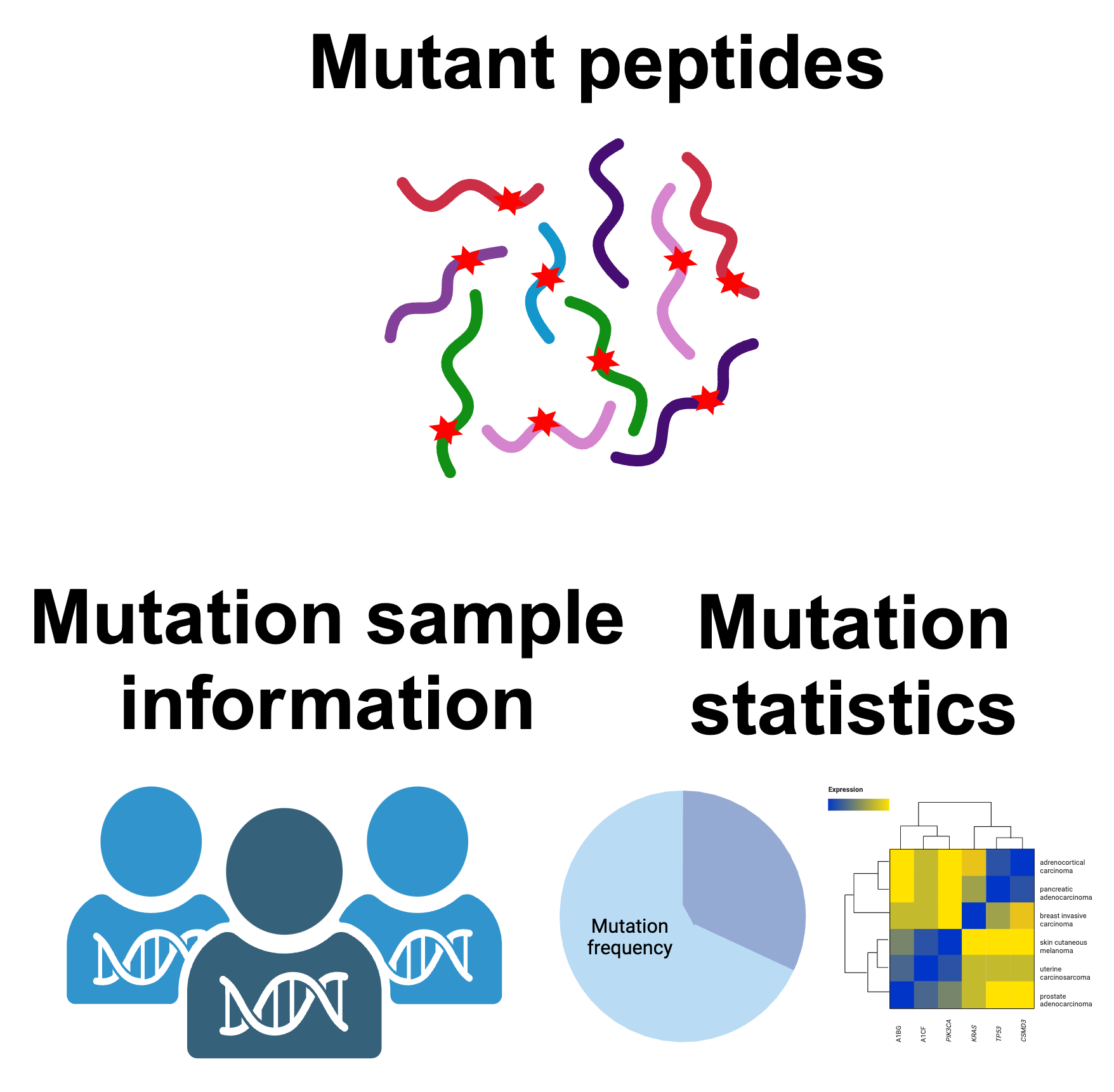
Extraction of mutant peptide sequences:
- Peptide Length: 25 amino acids (12 residues upstream + mutation + 12 residues downstream)
- Coverage: Sufficient for HLA class I peptides (8-14 amino acids)
- Output: 1,194,608 unique mutant peptides from 19,768 genes
Libraries optimized for MS search engines (FragPipe, PEAKS, DIA-NN)
-
CAN-IMMUNE Platform
Web-based interface and API

Comprehensive web platform features:
- Browse: Explore mutations by cell line, tissue, or cancer type
- Search: Query and filter mutations with Elasticsearch
- Statistics: Interactive visualizations of mutation distributions
- Download: Export libraries in multiple formats
- MutPep Tool: Generate custom libraries from user data
Access CAN-IMMUNE database functions -
LC-MS/MS Immunopeptidomics Analysis
Mass spectrometry-based identification
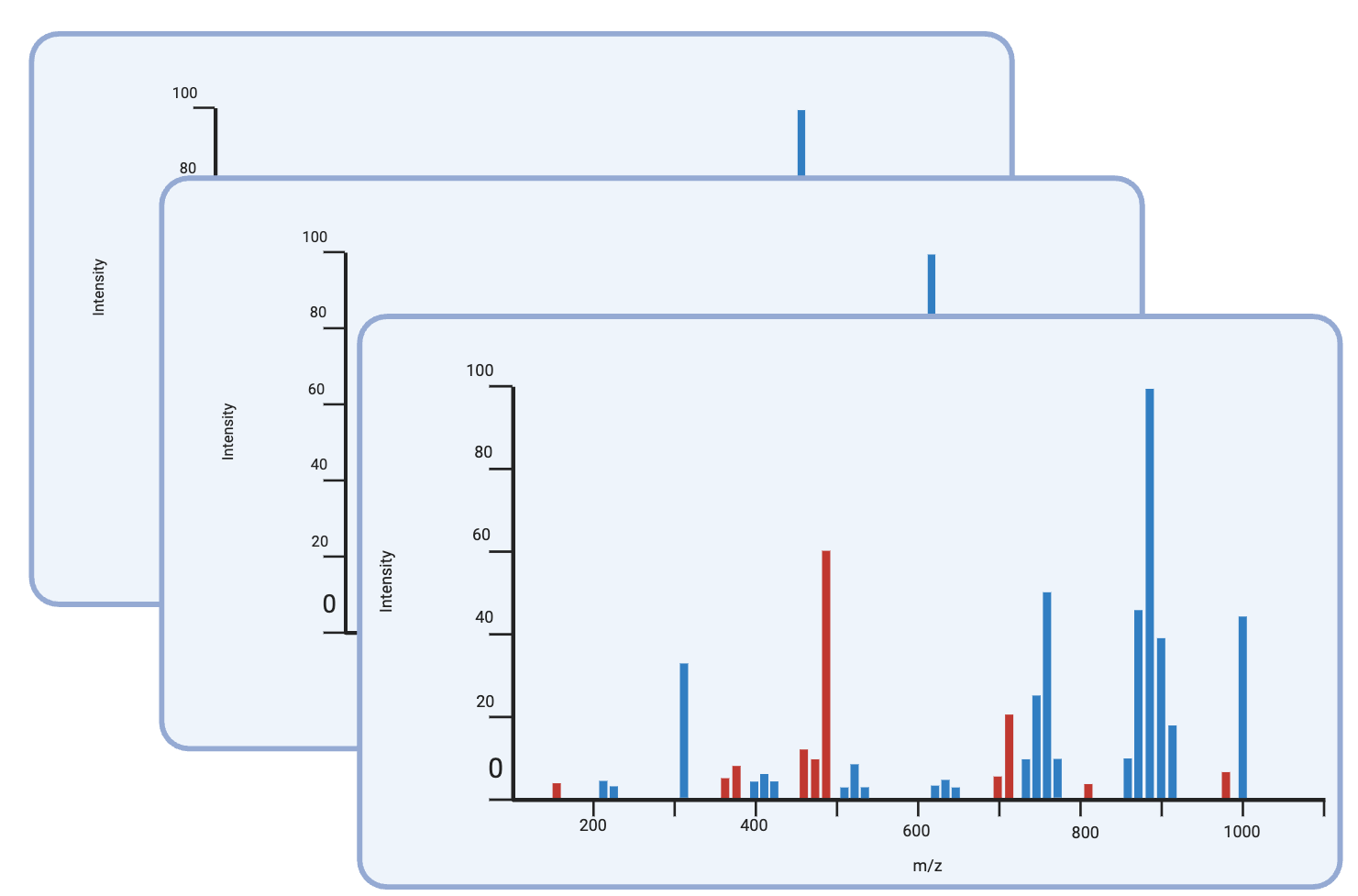
Comprehensive MS workflow:
- Sample Preparation: MHC peptide purification from cancer cells
- LC-MS/MS Acquisition: High-resolution mass spectrometry
- Database Search: MSFragger/PEAKS with custom mutant libraries
- FDR Control: Stringent 1% false discovery rate
Compatible with multiple search platforms for neoantigen discovery
-
Peptide Rescoring & Validation
Enhanced confidence scoring

Advanced rescoring algorithms:
- Percolator: Statistical FDR control
- MSBooster: Deep learning-based features
- MS2Rescore: Peptide identification enhancement
- PeptideProphet: Probability scoring (>0.9 threshold)
Reduces false positives in expanded search spaces
-
HLA Binding Affinity Prediction
NetMHCpan 4.1 analysis
Peptide-HLA binding classification:
- Strong Binders: %EL_rank ≤ 0.5
- Weak Binders: 0.5 < %EL_rank ≤ 2
- Non-Binders: %EL_rank > 2
Supports multiple HLA alleles (Class I: HLA-A, -B, -C)
Case Study Results: 76% of TNBC mutant peptides predicted as strong binders
-
Structural Modeling & TCR Interaction
AI-powered structure prediction

Advanced computational modeling:
- PANDORA: Fast peptide-HLA complex modeling
- AlphaFold2: TCR:peptide-MHC structure prediction
- Analysis: Binding energy and conformational assessment
- Comparison: Mutant vs. wild-type structural differences
Identifies structural alterations affecting T-cell recognition
-
Neoantigen Candidate Prioritization
Ranking for experimental validation
Multi-criteria ranking system:
- Peptide Quality: PeptideProphet score > 0.9
- HLA Binding: Strong binder classification
- Structural Stability: Optimal peptide-HLA conformation
- TCR Recognition: Favorable interaction interfaces
- Expression: RNA-seq validation (optional)
Output: Ranked list of high-confidence neoantigen candidates for immunogenicity testing