CAN-IMMUNE Tools
MutPep: A Key Toolkit of CAN-IMMUNE for Generating Mutant Peptide Libraries
View on GitHubOverview
MutPep is a Python-based standalone tool and a key component of CAN-IMMUNE that enables researchers to generate bespoke mutant peptide libraries from various mutation data sources. It streamlines the process of generating mass spectrometry-compatible libraries for cancer neoantigen discovery.
Key Features
- Multi-source data support (VCF, MAF, COSMIC)
- RefSeq GRCh38 reference database
- Parallel processing for large datasets
- Multiple output formats (FASTA, metadata, HTML)
- User-friendly GUI interface
- Comprehensive statistical reports
Quick Stats
Processing Speed:
~1 min for 3,455 mutations
Peptide Length:
25 amino acids (customizable)
System Requirements:
8-core CPU, 16GB RAM
MutPep Workflow
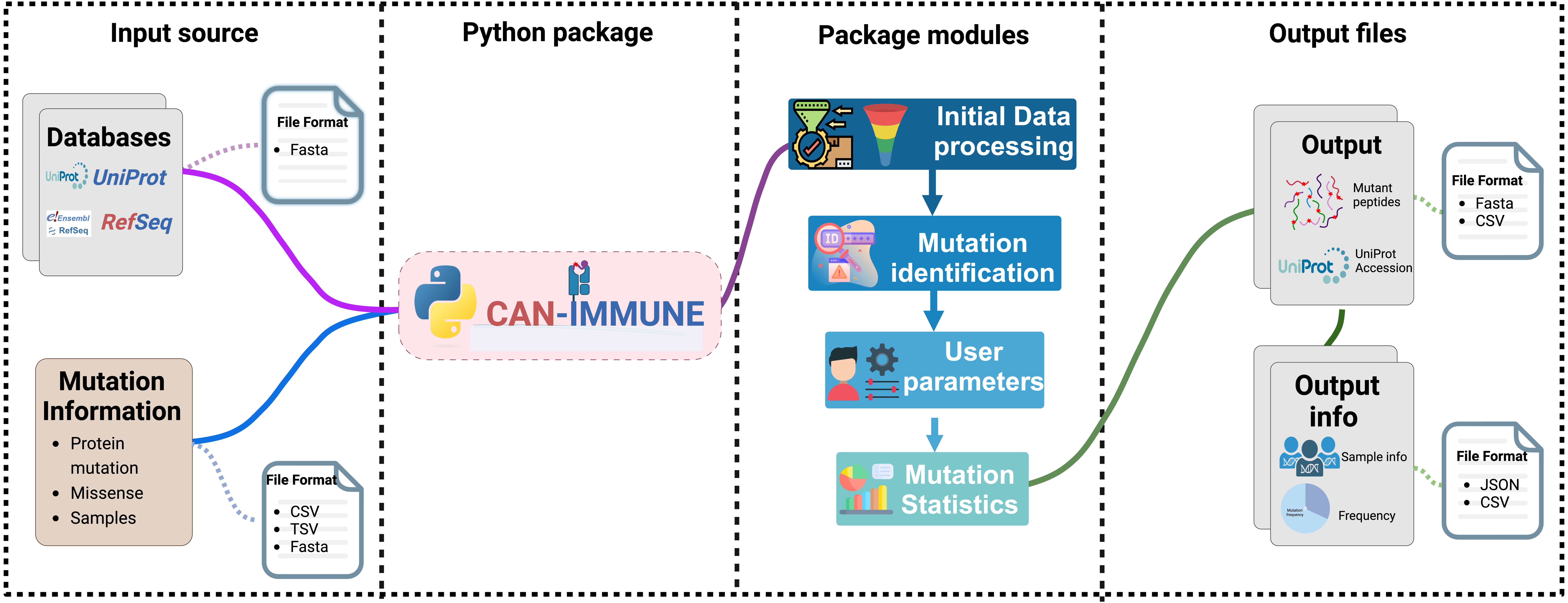
The MutPep workflow consists of four main steps that transform raw mutation data into searchable peptide libraries compatible with major mass spectrometry search engines:
Step 1: Data Source Processing
Supported Input Formats:
- VCF Files: Variant Call Format from sequencing pipelines
- MAF Files: Mutation Annotation Format from TCGA/GDC
- COSMIC Data: Direct integration with COSMIC database
- Custom Lists: User-defined mutation tables (CSV/TSV)
MutPep specifically processes missense mutations, which are most relevant for neoantigen discovery.
Step 2: Mutation Validation & Mapping
Cross-referencing Process:
- Validates mutation annotations against RefSeq protein database (GRCh38)
- Verifies wildtype residue, position, and mutant residue (e.g., p.A80P)
- Maps to correct protein transcript IDs
- Generates 25-amino acid peptides (12 residues flanking each side)
The 25-amino acid length ensures coverage of HLA class I peptides (typically 8-14 amino acids).
Step 3: Statistical Analysis
Generated Statistics:
- Most frequent mutant amino acids
- Mutant peptide length distribution
- Valid vs. invalid transcript ID analysis
- Success rate of mutation mapping
- Processing performance metrics
Step 4: Output Generation
Three Output Types:
FASTA Format
Mutant peptide libraries optimized for FragPipe, PEAKS, DIA-NN
Metadata Files
Input mappings and transcript IDs for reference
HTML Reports
Interactive data tables and processing statistics
Example Use Case: TCGA Breast Cancer Data
Input Data
Source: GDC Data Portal
Total Mutations: 3,455 missense mutations
Processing Time: ~1 minute
Processing Results
- Successfully Mapped: 3,245 mutations (93.9%)
- Failed (Non-missense): 210 entries
- Unmapped Transcripts: 210 IDs (version discrepancies)
Top Mutations Identified
| Substitution | Count | Percentage |
|---|---|---|
| Lysine (K) | 342 | 10.5% |
| Glutamine (Q) | 298 | 9.2% |
| Asparagine (N) | 276 | 8.5% |
Multi-threading enabled faster processing on standard hardware (8-core CPU, 16GB RAM)
Installation
Requirements:
- Python 3.8+
- pandas, numpy, BioPython
- RefSeq database (GRCh38)
Install via pip:
pip install mutpep
Or clone from GitHub:
git clone https://github.com/sanjaysgk/CanImmune.git
Basic Usage
Command Line:
mutpep --input mutations.maf \
--reference refseq_grch38.fasta \
--output output_dir \
--peptide-length 25
Python API:
from mutpep import MutPepGenerator
generator = MutPepGenerator()
generator.process_mutations('input.maf')
generator.generate_library()